








乳腺癌作为全球女性最常见的恶性肿瘤之一,其分子异质性和复杂的基因组特征一直是精准治疗的重要挑战。
近日,由韩国三星医疗中心(Samsung Medical Center)、首尔圣玛丽医院(Seoul St. Mary's Hospital)和生物信息学公司Inocras组成的研究团队在《自然》期刊发表了迄今为止规模最大的乳腺癌全基因组测序研究。

(来源:Nature)
该研究对1364例乳腺癌患者进行了全基因组测序(WGS)并结合完整的临床数据,鉴定出超过1090万个体细胞突变,发现了同源重组修复缺陷(HRD)在不同治疗情境下的预测价值,并揭示肿瘤基因组不稳定性可追溯至青春期早期。
研究通讯作者、三星医疗中心Yeon Hee Park教授指出:“整合基因组数据与详细的临床结果,为更个体化和有效的治疗策略铺平了道路,最终目标是改善患者预后。”
乳腺癌根据基因表达谱可分为管腔A型(luminal A)、管腔B型(luminal B)、HER2富集型(HER2-enriched)、基底样型(basal-like)和正常样型(normal-like)五种分子亚型,不同亚型的治疗策略和预后存在显著差异。然而,复发和转移仍然是临床面临的主要挑战,这凸显了深入理解乳腺癌基因组特征的迫切性。
传统的基因组研究主要依赖靶向测序技术,这类方法虽然聚焦于已知癌基因的个体突变,却遗漏了大量靶点以外的重要信息。基因组重排、拷贝数变异(CNA)和突变特征等模式驱动的基因组特征,往往无法被传统方法捕获。
相比之下,全基因组测序作为一种更全面的技术,能够捕获基因组变化的全谱,提供癌症基因组的无偏见视图,为生物学发现和潜在生物标志物的探索开辟了新的可能性。
尽管过去几十年学术界已对相当多的癌症基因组进行了分析,但这些研究的临床意义往往因缺乏与临床记录的充分整合而受限。要真正发挥基因组测序的实际价值,关键在于将基因组数据与全面的医疗记录有机结合,涵盖治疗反应、疾病复发和长期临床结果等多个维度。此外,既往乳腺癌基因组研究还面临样本量相对有限的制约,这限制了对低频突变和亚型特异性变异的深入挖掘。
值得关注的是,东西方人群在乳腺癌的分子特征上存在明显差异。韩国等东亚人群的乳腺癌患者普遍年龄更轻、雌激素受体阳性(ER+)比例更低,这使得针对亚洲人群的大规模基因组研究具有独特的科学价值和临床意义。
本研究构建的CUBRICS队列恰好填补了这一空白,作为迄今最大的整合全基因组测序和完整临床数据的乳腺癌研究队列,其患者中位年龄仅为44岁,显著低于西方国家,为探索这一独特人群的基因组特征提供了理想的研究平台。研究团队采用Inocras公司专有的CancerVision平台对所有样本进行分析,该平台不仅能够高效处理肿瘤-正常配对样本,实现高临床准确度和可扩展性,更支持原始全基因组测序数据与精心整理的临床记录的深度整合,为后续分析奠定了坚实基础。
该研究纳入来自三星医疗中心和首尔圣玛丽医院的1364例乳腺癌患者,在2012年至2023年间通过前瞻性和回顾性队列招募。88.6%的病例(1209例)同步进行了转录组测序,使研究团队能够将癌症分层为五种PAM50亚型,并追踪获得性基因组变异的表达。
通过全基因组测序,研究团队鉴定出10929118个体细胞突变,包括8935132个单核苷酸变异(SNVs)、1785446个插入缺失突变(Indels)和208540个结构变异(SVs),肿瘤突变负荷(TMB)的中位数为4742个突变。研究应用IntOGen流程鉴定出41个乳腺癌驱动基因,包括4个新发现的候选基因,如BCL11B基因在23例患者中发生突变,频率显著高于随机预期。
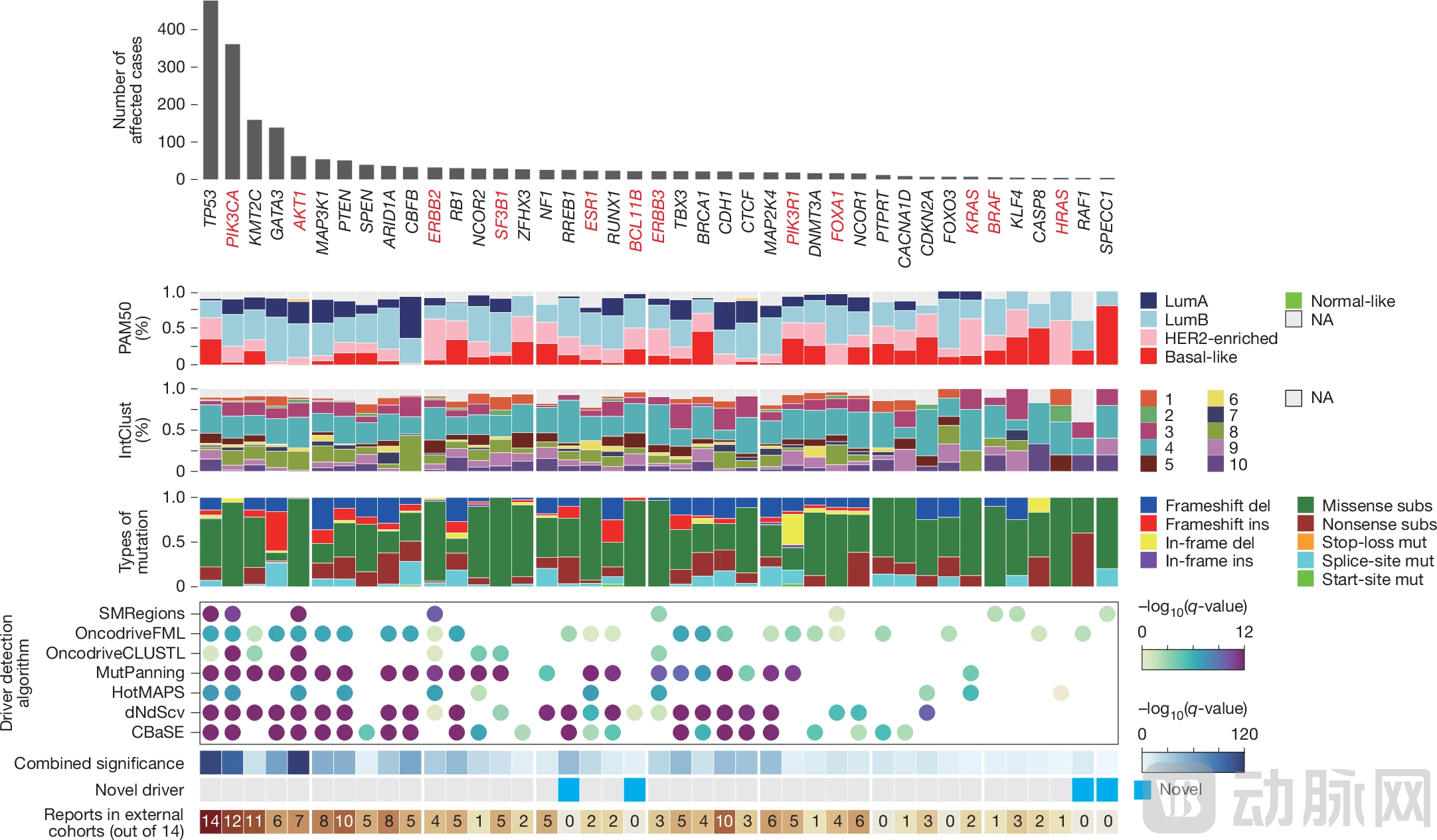
图:乳腺癌中的驱动基因(来源:Nature)
突变特征分析鉴定出17个单核苷酸变异特征、9个插入缺失特征和6个结构变异特征。与HRD相关的突变特征(SBS3、SBS8、ID6、SV3和SV5)尤为重要,它们是潜在的PARP抑制剂治疗反应预测标志物。
HRD在不同治疗情境下展现出截然相反的预后作用。在89例接受蒽环类-环磷酰胺辅助化疗的三阴性乳腺癌(TNBC)患者中,HRD患者(66例)的无病生存期显著优于同源重组修复正常(HRP)患者,风险比为0.10,验证了HRD肿瘤对DNA损伤类化疗的高敏感性。然而,在57例接受CDK4/6抑制剂联合内分泌治疗的激素受体阳性晚期乳腺癌患者中,13例HRD患者中85%出现疾病进展,无进展生存期显著短于HRP患者,风险比为4.20。多因素Cox回归分析显示,HRD是预测该治疗方案无进展生存期的最显著因素,风险比达10.20。
APOBEC相关突变特征在633个样本中贡献了超过10%的体细胞单核苷酸变异。APOBEC3A和APOBEC3B的胚系缺失在该队列中的等位基因频率为31.8%(1364例中736例),显著高于欧洲人群的8.5%(P<0.001),提示该变异在东亚人群中富集。携带该缺失的患者表现出更高的TMB(中位数5148 vs 4325,P<0.001)。
结构变异分析发现,15例乳腺癌携带8号和11号染色体间易位,将CCND1与ZNF703/FGFR1基因座置于近邻,可能通过增强子劫持机制促进癌基因表达。管腔型乳腺癌中频发融合包括MIPOL1-TTC6(9例)、CEP112-PRKCA(6例)和CCDC170-ESR1(6例),基底样型中则以BCL2L14-ETV6(12例)、AGO2-PTK2(6例)和BRD4-NOTCH3(6例)为主。CCDC170-ESR1融合已被证实与内分泌治疗耐药和转移相关。
时间分析揭示,大多数复发性长片段拷贝数扩增模式在癌症最近共同祖先细胞出现时就已获得,比临床诊断早数十年。研究者指出:“这意味着长片段拷贝数扩增是乳腺癌早期演化事件,推测可能早在青春期早期就已发生。”这一发现提示,从最初基因组不稳定事件到完整恶性转化可能需要数十年时间。
此外,染色体外DNA(ecDNA)驱动的ERBB2扩增显示出独特预测价值。在TransNEO队列的168例HER2阳性乳腺癌患者中,所有不携带ERBB2局灶扩增的患者均未达到新辅助化疗的病理完全缓解,而携带局灶扩增的4例患者中3例(75%)获得了完全缓解。
该研究揭示了全基因组测序在推进乳腺癌精准肿瘤学方面的潜力,为临床决策提供了重要启示。
HRD作为预测性生物标志物展现出治疗情境依赖性。在三阴性乳腺癌辅助化疗中,HRD预示更好的预后,符合DNA损伤修复缺陷肿瘤对化疗高敏感的认知;但在激素受体阳性晚期乳腺癌的CDK4/6抑制剂治疗中,HRD却与更差的预后相关。这种情境依赖性强调了在特定临床背景下精准评估生物标志物的重要性。
肿瘤异质性的定量评估显示出临床价值。肿瘤突变等位基因异质性(MATH)评分与总生存期相关,特别是在TP53突变的肿瘤中,高MATH评分与更差的预后相关。全基因组测序相比传统病理评估,能够提供更全面的肿瘤异质性刻画,捕获亚克隆突变带来的遗传多样性。
通过拷贝数变异的时间分析,研究团队重建了癌症演化的时间轨迹,揭示基因组不稳定性在肿瘤诊断前数十年就已发生。这一发现将基因组研究从传统的“二维平面”拓展到“三维时空”,为理解癌症生物学、肿瘤发生的多阶段演化过程,以及治疗过程中耐药机制的动态演变提供了新视角。
种族差异发现具有重要意义。APOBEC3A/B缺失在韩国人群中的频率(31.8%)远超欧洲人群(8.5%),且与更高的肿瘤突变负荷相关。这提示不同种族人群可能因遗传背景差异而面临不同的基因组风险模式,需要发展差异化的风险评估策略和个体化治疗方案。
研究证明了全基因组测序结合真实世界临床数据的新范式。通过整合大规模全基因组数据与临床记录,在回顾性分析中实现了数据驱动的生物标志物发现,降低了研究成本和时间周期,为加速生物标志物临床转化提供了新路径。
展望未来,前瞻性临床试验对于验证这些基因组改变的功能意义仍然重要。基于全基因组测序的肿瘤异质性定量评估,将在塑造未来精准肿瘤学策略中发挥核心作用。随着测序成本下降、分析技术成熟,以及人工智能在基因组数据解读中的应用,全基因组测序整合到常规癌症诊疗的前景日益清晰。该研究建立的包含1364例乳腺癌全基因组和临床数据的资源,将成为未来癌症基因组智能平台的参考数据集。
这项最大规模的乳腺癌全基因组研究,通过整合超过1090万个突变与详细临床数据,绘制了乳腺癌基因组演化的全景图谱。从青春期早期的基因组不稳定事件到数十年后的肿瘤诊断,从HRD在不同治疗情境下的双重作用到染色体外DNA驱动的治疗反应,这些发现深化了对乳腺癌分子机制的理解,为精准肿瘤学发展奠定了基础。随着全基因组测序技术的普及和多组学数据的整合,基于患者分子特征的个体化治疗将从愿景走向现实,最终改善乳腺癌患者的预后。


















